Adsorption.avi (4.73 Mb)
Top
Microscopic simulation of deposition and nucleation of silicon nano particles at the silicon oxide surface
The results of calculation have shown the maximal bond energy on crystal and on amorphous silicon oxide surface is realized when silicon atom interacts with two oxygen atoms i.e. when O-Si-O structure is arisen. The distribution function of silicon atom bond energy on crystal and amorphous silicon oxide surface was obtained too using molecular - dynamics simulation technique.
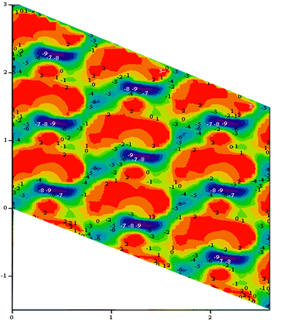
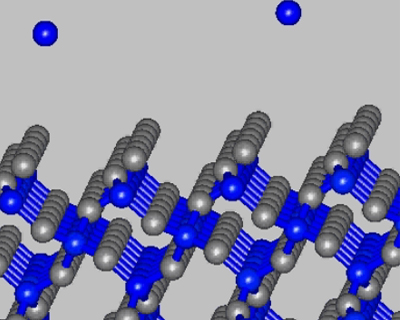
Figure 1 Crystal SiO2 surface
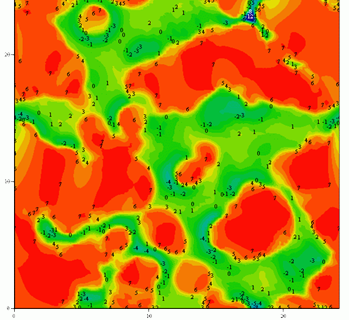
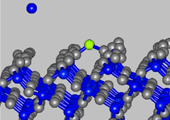
Figure 2 Amorphous SiO2 surface
Top
The bond energy Isosurface for the crystal and amorphous SiO2 surface
The method of near – surface layer scanning was used to calculate isoenergetic surfaces for the bond energy field of silicon atom in the near – surface layers of crystal and amorphous silicon oxide. Surface density of attraction areas for silicon atom was calculated on surface (001) of crystal and amorphous silicon oxide. We calculated the surface density value of silicon atoms localization places. This value was compared with experimental surface density of silicon nanoparticles.
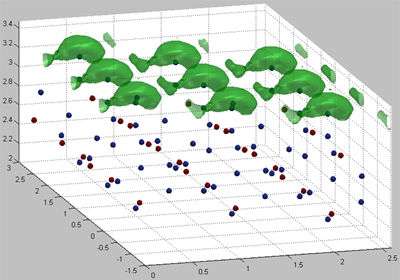
Figure 3 The value on this isosurface is -5 eV
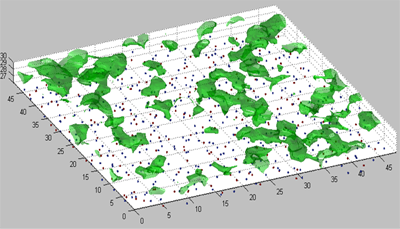
Figure 4 The value on this isosurface is -2 eV
Top